Portfolio
Bioinformatics Lab Course @ UCSD
class13
Pooja Parthasarathy | A17817456
Background
Today we will perform an RNASeq analysis on the effects of dexamethasone (dex for short), a common steroid on airway smooth muscle (ASM) cell lines
Data
We need 2 things for this analysis - count and col data - countData: the biggest of the two, the table with genes as rows and samples as columns - **colData*: the metadata about the columns (i.e, sanples) in the main countData object
counts <- read.csv("airway_scaledcounts.csv", row.names = 1)
metadata <- read.csv("airway_metadata.csv")
(metadata)
id dex celltype geo_id
1 SRR1039508 control N61311 GSM1275862
2 SRR1039509 treated N61311 GSM1275863
3 SRR1039512 control N052611 GSM1275866
4 SRR1039513 treated N052611 GSM1275867
5 SRR1039516 control N080611 GSM1275870
6 SRR1039517 treated N080611 GSM1275871
7 SRR1039520 control N061011 GSM1275874
8 SRR1039521 treated N061011 GSM1275875
head(counts)
SRR1039508 SRR1039509 SRR1039512 SRR1039513 SRR1039516
ENSG00000000003 723 486 904 445 1170
ENSG00000000005 0 0 0 0 0
ENSG00000000419 467 523 616 371 582
ENSG00000000457 347 258 364 237 318
ENSG00000000460 96 81 73 66 118
ENSG00000000938 0 0 1 0 2
SRR1039517 SRR1039520 SRR1039521
ENSG00000000003 1097 806 604
ENSG00000000005 0 0 0
ENSG00000000419 781 417 509
ENSG00000000457 447 330 324
ENSG00000000460 94 102 74
ENSG00000000938 0 0 0
Check on metadata count corespondance
We need to check that the metadata matches the samples in our count data.
ncol(counts) ==
nrow(metadata)
[1] TRUE
all(colnames(counts) ==
metadata$id)
[1] TRUE
Q1.How many genes in this dataset
nrow(counts)
[1] 38694
Q2. How many control samples are in this dataset
sum(metadata$dex == "control")
[1] 4
Analysis Plan
Q3 We have 4 replicates per condition (cotnrol & treated), we want to compare the control vs the treated to see which genes expression levels change wen we have the drug present.
To do this you first group by celltype, find a summary statistic to represent the average expression level for the gene
- Step 1. find which samples of columns in counts correspond to “control” samples.
- Step 2. Exreact/select these control columns
- Step 3. Across these control samples generate an aveage for each gene’s expression (utilize MEAN ACROSS ROWS)
#the indices (i.e positions) that are "control"
control.inds <- metadata$dex == "control"
control.counts <- counts[, control.inds]
#calculate the mean for each gene (i.e row)
control.mean <- rowMeans(control.counts)
Now let us do the same for treated samples
treated.inds <- metadata$dex == "treated"
treated.counts <- counts[, treated.inds]
treated.mean <- rowMeans(treated.counts)
Let’s put these two mean values in a new data.frame for easy book keeping and plotting
meancounts <- data.frame(control.mean, treated.mean)
head(meancounts)
control.mean treated.mean
ENSG00000000003 900.75 658.00
ENSG00000000005 0.00 0.00
ENSG00000000419 520.50 546.00
ENSG00000000457 339.75 316.50
ENSG00000000460 97.25 78.75
ENSG00000000938 0.75 0.00
Q5
library(ggplot2)
ggplot(meancounts) +
aes(control.mean, treated.mean) +
geom_point(alpha = 0.3)
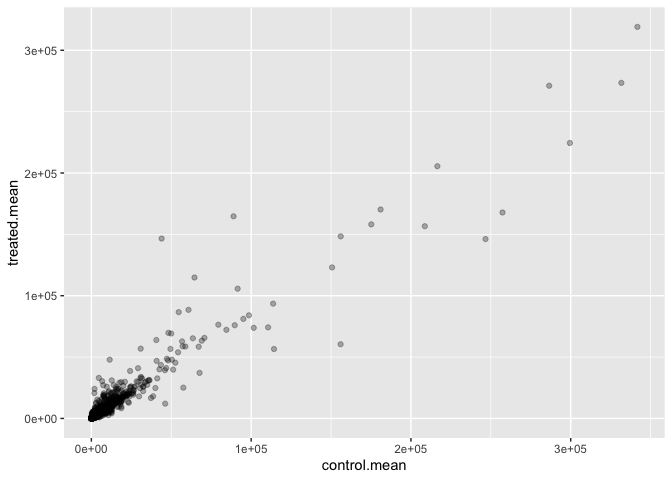
Make a ggplot of average counts of control versus treated
ggplot(meancounts) +
aes(control.mean, treated.mean) +
geom_point(alpha = 0.3) +
scale_x_log10() +
scale_y_log10()
Warning in scale_x_log10(): log-10 transformation introduced infinite values.
Warning in scale_y_log10(): log-10 transformation introduced infinite values.
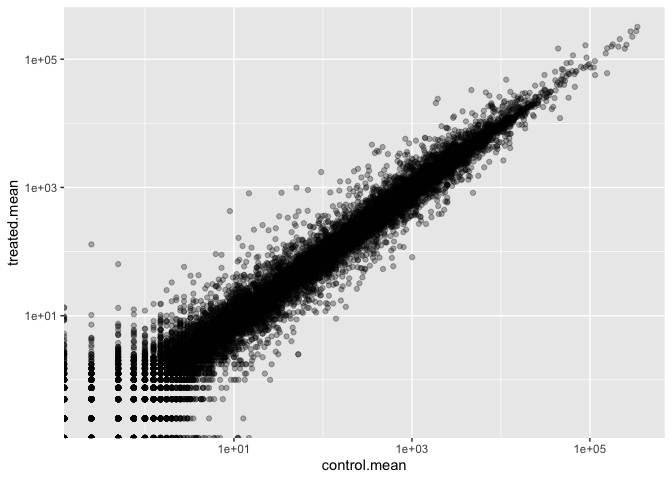
Log2 units and fold change
If we consider treated/control counts we will get a number that tells us the change (fold change) is the log of this fraction meaning for instange log(1/1) = 0 therefore no hange while the fraction itself may equal 1. A doublign will lead to +1 log(2) = 1, halving will be -1.
idea - take existing mean counts data frame and make an additional fold change column
log2(10/20)
[1] -1
Add a new column
log2fcfor log2 fold change of treated/control to ourmeancountsobject
meancounts$log2fc <- log2(meancounts$treated.mean/meancounts$control.mean)
head(meancounts)
control.mean treated.mean log2fc
ENSG00000000003 900.75 658.00 -0.45303916
ENSG00000000005 0.00 0.00 NaN
ENSG00000000419 520.50 546.00 0.06900279
ENSG00000000457 339.75 316.50 -0.10226805
ENSG00000000460 97.25 78.75 -0.30441833
ENSG00000000938 0.75 0.00 -Inf
zero.vals <- which(meancounts[,1:2]==0, arr.ind=TRUE)
to.rm <- unique(zero.vals[,1])
mycounts <- meancounts[-to.rm,]
head(mycounts)
control.mean treated.mean log2fc
ENSG00000000003 900.75 658.00 -0.45303916
ENSG00000000419 520.50 546.00 0.06900279
ENSG00000000457 339.75 316.50 -0.10226805
ENSG00000000460 97.25 78.75 -0.30441833
ENSG00000000971 5219.00 6687.50 0.35769358
ENSG00000001036 2327.00 1785.75 -0.38194109
Q7. What is the purpose of the arr.ind argument in the which() function call above? Why would we then take the first column of the output and need to call the unique() function?
The arr.ind=TRUE helps find indices aka rows and columns where it is TRUE, or for us - having 0 counts. Calling unique() will ensure we don’t count any row twice if it has zero entries in both samples.
Now we can use some fold thresholds to just check for up and downregulation
up.ind <- mycounts$log2fc > 2
down.ind <- mycounts$log2fc < (-2)
Q8
sum(up.ind)
[1] 250
Q9
sum(down.ind)
[1] 367
Q10
We cannot fully tell as the results have not been tested for their significance yet!
Setting up DESeq2
Let’s do this the right way. DESeq2 is an R package specifically for analyzing count-based NGS data like RNA-seq. It is available from Bioconductor. Bioconductor is a project to provide tools for analyzing high-throughput genomic data including RNA-seq, ChIP-seq and arrays. You can explore Bioconductor packages here.
Bioconductor packages usually have great documentation in the form of vignettes. For a great example, take a look at the DESeq2 vignette for analyzing count data. This 40+ page manual is packed full of examples on using DESeq2, importing data, fitting models, creating visualizations, references, etc.
Just like R packages from CRAN, you only need to install Bioconductor packages once (instructions here), then load them every time you start a new R session.
# Load up DESeq2
library(DESeq2)
Loading required package: S4Vectors
Loading required package: stats4
Loading required package: BiocGenerics
Loading required package: generics
Attaching package: 'generics'
The following objects are masked from 'package:base':
as.difftime, as.factor, as.ordered, intersect, is.element, setdiff,
setequal, union
Attaching package: 'BiocGenerics'
The following objects are masked from 'package:stats':
IQR, mad, sd, var, xtabs
The following objects are masked from 'package:base':
anyDuplicated, aperm, append, as.data.frame, basename, cbind,
colnames, dirname, do.call, duplicated, eval, evalq, Filter, Find,
get, grep, grepl, is.unsorted, lapply, Map, mapply, match, mget,
order, paste, pmax, pmax.int, pmin, pmin.int, Position, rank,
rbind, Reduce, rownames, sapply, saveRDS, table, tapply, unique,
unsplit, which.max, which.min
Attaching package: 'S4Vectors'
The following object is masked from 'package:utils':
findMatches
The following objects are masked from 'package:base':
expand.grid, I, unname
Loading required package: IRanges
Loading required package: GenomicRanges
Loading required package: Seqinfo
Loading required package: SummarizedExperiment
Loading required package: MatrixGenerics
Loading required package: matrixStats
Attaching package: 'MatrixGenerics'
The following objects are masked from 'package:matrixStats':
colAlls, colAnyNAs, colAnys, colAvgsPerRowSet, colCollapse,
colCounts, colCummaxs, colCummins, colCumprods, colCumsums,
colDiffs, colIQRDiffs, colIQRs, colLogSumExps, colMadDiffs,
colMads, colMaxs, colMeans2, colMedians, colMins, colOrderStats,
colProds, colQuantiles, colRanges, colRanks, colSdDiffs, colSds,
colSums2, colTabulates, colVarDiffs, colVars, colWeightedMads,
colWeightedMeans, colWeightedMedians, colWeightedSds,
colWeightedVars, rowAlls, rowAnyNAs, rowAnys, rowAvgsPerColSet,
rowCollapse, rowCounts, rowCummaxs, rowCummins, rowCumprods,
rowCumsums, rowDiffs, rowIQRDiffs, rowIQRs, rowLogSumExps,
rowMadDiffs, rowMads, rowMaxs, rowMeans2, rowMedians, rowMins,
rowOrderStats, rowProds, rowQuantiles, rowRanges, rowRanks,
rowSdDiffs, rowSds, rowSums2, rowTabulates, rowVarDiffs, rowVars,
rowWeightedMads, rowWeightedMeans, rowWeightedMedians,
rowWeightedSds, rowWeightedVars
Loading required package: Biobase
Welcome to Bioconductor
Vignettes contain introductory material; view with
'browseVignettes()'. To cite Bioconductor, see
'citation("Biobase")', and for packages 'citation("pkgname")'.
Attaching package: 'Biobase'
The following object is masked from 'package:MatrixGenerics':
rowMedians
The following objects are masked from 'package:matrixStats':
anyMissing, rowMedians
dds <- DESeqDataSetFromMatrix(countData=counts,
colData=metadata,
design=~dex)
converting counts to integer mode
Warning in DESeqDataSet(se, design = design, ignoreRank): some variables in
design formula are characters, converting to factors
dds <- DESeq(dds)
estimating size factors
estimating dispersions
gene-wise dispersion estimates
mean-dispersion relationship
final dispersion estimates
fitting model and testing
res <- results(dds)
res
log2 fold change (MLE): dex treated vs control
Wald test p-value: dex treated vs control
DataFrame with 38694 rows and 6 columns
baseMean log2FoldChange lfcSE stat pvalue
<numeric> <numeric> <numeric> <numeric> <numeric>
ENSG00000000003 747.1942 -0.350703 0.168242 -2.084514 0.0371134
ENSG00000000005 0.0000 NA NA NA NA
ENSG00000000419 520.1342 0.206107 0.101042 2.039828 0.0413675
ENSG00000000457 322.6648 0.024527 0.145134 0.168996 0.8658000
ENSG00000000460 87.6826 -0.147143 0.256995 -0.572550 0.5669497
... ... ... ... ... ...
ENSG00000283115 0.000000 NA NA NA NA
ENSG00000283116 0.000000 NA NA NA NA
ENSG00000283119 0.000000 NA NA NA NA
ENSG00000283120 0.974916 -0.66825 1.69441 -0.394385 0.693297
ENSG00000283123 0.000000 NA NA NA NA
padj
<numeric>
ENSG00000000003 0.163017
ENSG00000000005 NA
ENSG00000000419 0.175937
ENSG00000000457 0.961682
ENSG00000000460 0.815805
... ...
ENSG00000283115 NA
ENSG00000283116 NA
ENSG00000283119 NA
ENSG00000283120 NA
ENSG00000283123 NA
We can get some basic summaries tallies using the summary() function
summary(res, alpha = 0.05)
out of 25258 with nonzero total read count
adjusted p-value < 0.05
LFC > 0 (up) : 1242, 4.9%
LFC < 0 (down) : 939, 3.7%
outliers [1] : 142, 0.56%
low counts [2] : 9971, 39%
(mean count < 10)
[1] see 'cooksCutoff' argument of ?results
[2] see 'independentFiltering' argument of ?results
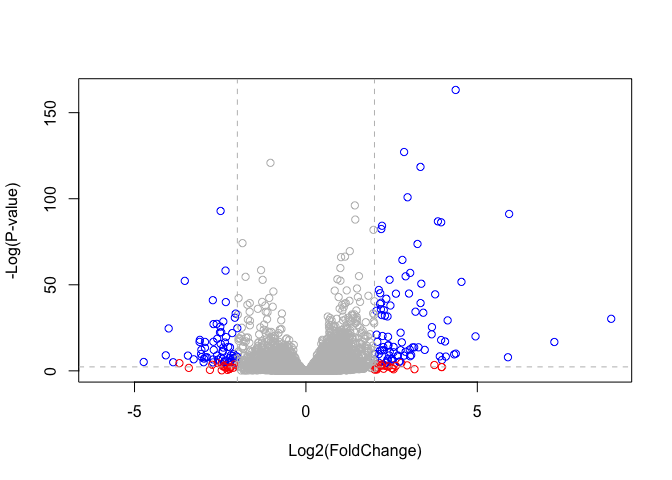
Data Visualization - Volcano plots
plot(res$log2FoldChange, res$padj)
plot(res$log2FoldChange, -log(res$padj))
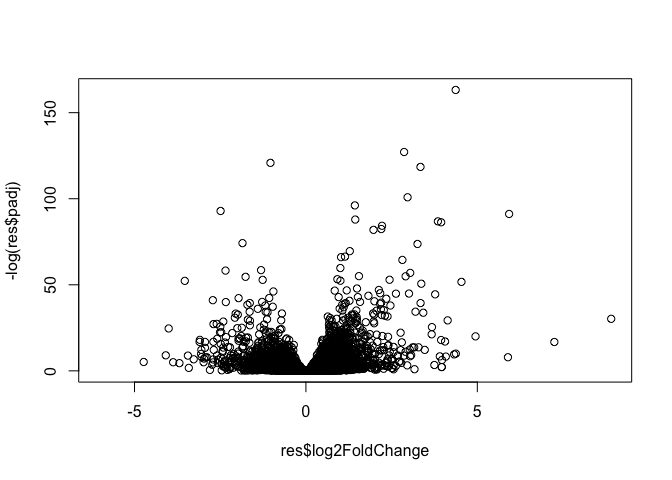
# Setup our custom point color vector
mycols <- rep("gray", nrow(res))
mycols[ abs(res$log2FoldChange) > 2 ] <- "red"
inds <- (res$padj < 0.01) & (abs(res$log2FoldChange) > 2 )
mycols[ inds ] <- "blue"
# Volcano plot with custom colors
plot( res$log2FoldChange, -log(res$padj),
col=mycols, ylab="-Log(P-value)", xlab="Log2(FoldChange)" )
# Cut-off lines
abline(v=c(-2,2), col="gray", lty=2)
abline(h=-log(0.1), col="gray", lty=2)