Portfolio
Bioinformatics Lab Course @ UCSD
class14 - RNA Seq mini project
Pooja Parthasarathy | PID: A17817456
pathway analysis helps connect using existing knowledge on pathways, how various gene sets to understand DEG - differentialy expression genes
project context - The authors report on differential analysis of lung fibroblasts in response to loss of the developmental transcription factor HOXA1. Their results and others indicate that HOXA1 is required for lung fibroblast and HeLa cell cycle progression. In particular their analysis show that “loss of HOXA1 results in significant expression level changes in thousands of individual transcripts, along with isoform switching events in key regulators of the cell cycle”. For our session we have used their Sailfish gene-level estimated counts and hence are restricted to protein-coding genes only.
##
library(DESeq2)
Loading required package: S4Vectors
Loading required package: stats4
Loading required package: BiocGenerics
Loading required package: generics
Attaching package: 'generics'
The following objects are masked from 'package:base':
as.difftime, as.factor, as.ordered, intersect, is.element, setdiff,
setequal, union
Attaching package: 'BiocGenerics'
The following objects are masked from 'package:stats':
IQR, mad, sd, var, xtabs
The following objects are masked from 'package:base':
anyDuplicated, aperm, append, as.data.frame, basename, cbind,
colnames, dirname, do.call, duplicated, eval, evalq, Filter, Find,
get, grep, grepl, is.unsorted, lapply, Map, mapply, match, mget,
order, paste, pmax, pmax.int, pmin, pmin.int, Position, rank,
rbind, Reduce, rownames, sapply, saveRDS, table, tapply, unique,
unsplit, which.max, which.min
Attaching package: 'S4Vectors'
The following object is masked from 'package:utils':
findMatches
The following objects are masked from 'package:base':
expand.grid, I, unname
Loading required package: IRanges
Loading required package: GenomicRanges
Loading required package: Seqinfo
Loading required package: SummarizedExperiment
Loading required package: MatrixGenerics
Loading required package: matrixStats
Attaching package: 'MatrixGenerics'
The following objects are masked from 'package:matrixStats':
colAlls, colAnyNAs, colAnys, colAvgsPerRowSet, colCollapse,
colCounts, colCummaxs, colCummins, colCumprods, colCumsums,
colDiffs, colIQRDiffs, colIQRs, colLogSumExps, colMadDiffs,
colMads, colMaxs, colMeans2, colMedians, colMins, colOrderStats,
colProds, colQuantiles, colRanges, colRanks, colSdDiffs, colSds,
colSums2, colTabulates, colVarDiffs, colVars, colWeightedMads,
colWeightedMeans, colWeightedMedians, colWeightedSds,
colWeightedVars, rowAlls, rowAnyNAs, rowAnys, rowAvgsPerColSet,
rowCollapse, rowCounts, rowCummaxs, rowCummins, rowCumprods,
rowCumsums, rowDiffs, rowIQRDiffs, rowIQRs, rowLogSumExps,
rowMadDiffs, rowMads, rowMaxs, rowMeans2, rowMedians, rowMins,
rowOrderStats, rowProds, rowQuantiles, rowRanges, rowRanks,
rowSdDiffs, rowSds, rowSums2, rowTabulates, rowVarDiffs, rowVars,
rowWeightedMads, rowWeightedMeans, rowWeightedMedians,
rowWeightedSds, rowWeightedVars
Loading required package: Biobase
Welcome to Bioconductor
Vignettes contain introductory material; view with
'browseVignettes()'. To cite Bioconductor, see
'citation("Biobase")', and for packages 'citation("pkgname")'.
Attaching package: 'Biobase'
The following object is masked from 'package:MatrixGenerics':
rowMedians
The following objects are masked from 'package:matrixStats':
anyMissing, rowMedians
metaFile <- "GSE37704_metadata.csv"
countFile <- "GSE37704_featurecounts.csv"
colData <- read.csv(metaFile, row.names = 1)
head(colData)
condition
SRR493366 control_sirna
SRR493367 control_sirna
SRR493368 control_sirna
SRR493369 hoxa1_kd
SRR493370 hoxa1_kd
SRR493371 hoxa1_kd
countData = read.csv(countFile, row.names=1)
head(countData)
length SRR493366 SRR493367 SRR493368 SRR493369 SRR493370
ENSG00000186092 918 0 0 0 0 0
ENSG00000279928 718 0 0 0 0 0
ENSG00000279457 1982 23 28 29 29 28
ENSG00000278566 939 0 0 0 0 0
ENSG00000273547 939 0 0 0 0 0
ENSG00000187634 3214 124 123 205 207 212
SRR493371
ENSG00000186092 0
ENSG00000279928 0
ENSG00000279457 46
ENSG00000278566 0
ENSG00000273547 0
ENSG00000187634 258
Q
# Note we need to remove the odd first $length col
countData <- as.matrix(countData[,-1])
head(countData)
SRR493366 SRR493367 SRR493368 SRR493369 SRR493370 SRR493371
ENSG00000186092 0 0 0 0 0 0
ENSG00000279928 0 0 0 0 0 0
ENSG00000279457 23 28 29 29 28 46
ENSG00000278566 0 0 0 0 0 0
ENSG00000273547 0 0 0 0 0 0
ENSG00000187634 124 123 205 207 212 258
Q. Complete the code below to filter countData to exclude genes (i.e. rows) where we have 0 read count across all samples (i.e. columns). Tip: What will rowSums() of countData return and how could you use it in this context?
countData = countData[rowSums(countData) > 0, ]
#nrow(countData)
head(countData)
SRR493366 SRR493367 SRR493368 SRR493369 SRR493370 SRR493371
ENSG00000279457 23 28 29 29 28 46
ENSG00000187634 124 123 205 207 212 258
ENSG00000188976 1637 1831 2383 1226 1326 1504
ENSG00000187961 120 153 180 236 255 357
ENSG00000187583 24 48 65 44 48 64
ENSG00000187642 4 9 16 14 16 16
dds = DESeqDataSetFromMatrix(countData=countData,
colData=colData,
design=~condition)
Warning in DESeqDataSet(se, design = design, ignoreRank): some variables in
design formula are characters, converting to factors
dds = DESeq(dds)
estimating size factors
estimating dispersions
gene-wise dispersion estimates
mean-dispersion relationship
final dispersion estimates
fitting model and testing
dds
class: DESeqDataSet
dim: 15975 6
metadata(1): version
assays(4): counts mu H cooks
rownames(15975): ENSG00000279457 ENSG00000187634 ... ENSG00000276345
ENSG00000271254
rowData names(22): baseMean baseVar ... deviance maxCooks
colnames(6): SRR493366 SRR493367 ... SRR493370 SRR493371
colData names(2): condition sizeFactor
res = results(dds)
summary(res)
out of 15975 with nonzero total read count
adjusted p-value < 0.1
LFC > 0 (up) : 4349, 27%
LFC < 0 (down) : 4396, 28%
outliers [1] : 0, 0%
low counts [2] : 1237, 7.7%
(mean count < 0)
[1] see 'cooksCutoff' argument of ?results
[2] see 'independentFiltering' argument of ?results
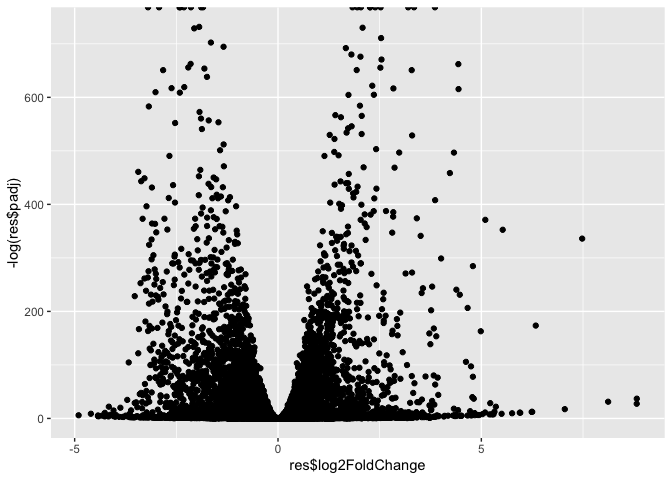
library(ggplot2)
ggplot(res) +
aes(res$log2FoldChange,
-log(res$padj)) +
geom_point()
Warning: Removed 1237 rows containing missing values or values outside the scale range
(`geom_point()`).
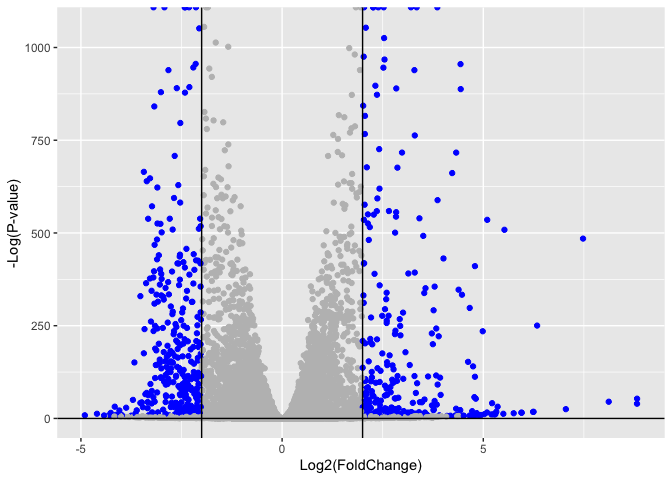
# Make a color vector for all genes
mycols <- rep("gray", nrow(res) )
# Color blue the genes with fold change above 2
mycols[ abs(res$log2FoldChange) > 2 ] <- "blue"
# Color gray those with adjusted p-value more than 0.01
mycols[ res$padj > 0.01 ] <- "gray"
ggplot(res) +
aes(res$log2FoldChange,
-log2(res$padj))+
geom_point(col=mycols) +
xlab("Log2(FoldChange)") +
ylab("-Log(P-value)") +
geom_vline(xintercept = c(-2,2)) +
geom_hline(yintercept = 0)
Warning: Removed 1237 rows containing missing values or values outside the scale range
(`geom_point()`).
library("AnnotationDbi")
library("org.Hs.eg.db")
library("AnnotationDbi")
library("org.Hs.eg.db")
columns(org.Hs.eg.db)
[1] "ACCNUM" "ALIAS" "ENSEMBL" "ENSEMBLPROT" "ENSEMBLTRANS"
[6] "ENTREZID" "ENZYME" "EVIDENCE" "EVIDENCEALL" "GENENAME"
[11] "GENETYPE" "GO" "GOALL" "IPI" "MAP"
[16] "OMIM" "ONTOLOGY" "ONTOLOGYALL" "PATH" "PFAM"
[21] "PMID" "PROSITE" "REFSEQ" "SYMBOL" "UCSCKG"
[26] "UNIPROT"
res$symbol = mapIds(org.Hs.eg.db,
keys=row.names(res),
keytype="ENSEMBL",
column= "SYMBOL",
multiVals="first")
'select()' returned 1:many mapping between keys and columns
res$entrez = mapIds(org.Hs.eg.db,
keys=row.names(res),
keytype="ENSEMBL",
column="ENTREZID",
multiVals="first")
'select()' returned 1:many mapping between keys and columns
res$name = mapIds(org.Hs.eg.db,
keys=row.names(res),
keytype="ENSEMBL",
column="GENENAME",
multiVals="first")
'select()' returned 1:many mapping between keys and columns
head(res, 10)
log2 fold change (MLE): condition hoxa1 kd vs control sirna
Wald test p-value: condition hoxa1 kd vs control sirna
DataFrame with 10 rows and 9 columns
baseMean log2FoldChange lfcSE stat pvalue
<numeric> <numeric> <numeric> <numeric> <numeric>
ENSG00000279457 29.913579 0.1792571 0.3248216 0.551863 5.81042e-01
ENSG00000187634 183.229650 0.4264571 0.1402658 3.040350 2.36304e-03
ENSG00000188976 1651.188076 -0.6927205 0.0548465 -12.630158 1.43990e-36
ENSG00000187961 209.637938 0.7297556 0.1318599 5.534326 3.12428e-08
ENSG00000187583 47.255123 0.0405765 0.2718928 0.149237 8.81366e-01
ENSG00000187642 11.979750 0.5428105 0.5215598 1.040744 2.97994e-01
ENSG00000188290 108.922128 2.0570638 0.1969053 10.446970 1.51282e-25
ENSG00000187608 350.716868 0.2573837 0.1027266 2.505522 1.22271e-02
ENSG00000188157 9128.439422 0.3899088 0.0467163 8.346304 7.04321e-17
ENSG00000237330 0.158192 0.7859552 4.0804729 0.192614 8.47261e-01
padj symbol entrez name
<numeric> <character> <character> <character>
ENSG00000279457 6.86555e-01 NA NA NA
ENSG00000187634 5.15718e-03 SAMD11 148398 sterile alpha motif ..
ENSG00000188976 1.76549e-35 NOC2L 26155 NOC2 like nucleolar ..
ENSG00000187961 1.13413e-07 KLHL17 339451 kelch like family me..
ENSG00000187583 9.19031e-01 PLEKHN1 84069 pleckstrin homology ..
ENSG00000187642 4.03379e-01 PERM1 84808 PPARGC1 and ESRR ind..
ENSG00000188290 1.30538e-24 HES4 57801 hes family bHLH tran..
ENSG00000187608 2.37452e-02 ISG15 9636 ISG15 ubiquitin like..
ENSG00000188157 4.21963e-16 AGRN 375790 agrin
ENSG00000237330 NA RNF223 401934 ring finger protein ..
res = res[order(res$pvalue),]
write.csv(res, file="deseq_results.csv")
library(pathview)
##############################################################################
Pathview is an open source software package distributed under GNU General
Public License version 3 (GPLv3). Details of GPLv3 is available at
http://www.gnu.org/licenses/gpl-3.0.html. Particullary, users are required to
formally cite the original Pathview paper (not just mention it) in publications
or products. For details, do citation("pathview") within R.
The pathview downloads and uses KEGG data. Non-academic uses may require a KEGG
license agreement (details at http://www.kegg.jp/kegg/legal.html).
##############################################################################
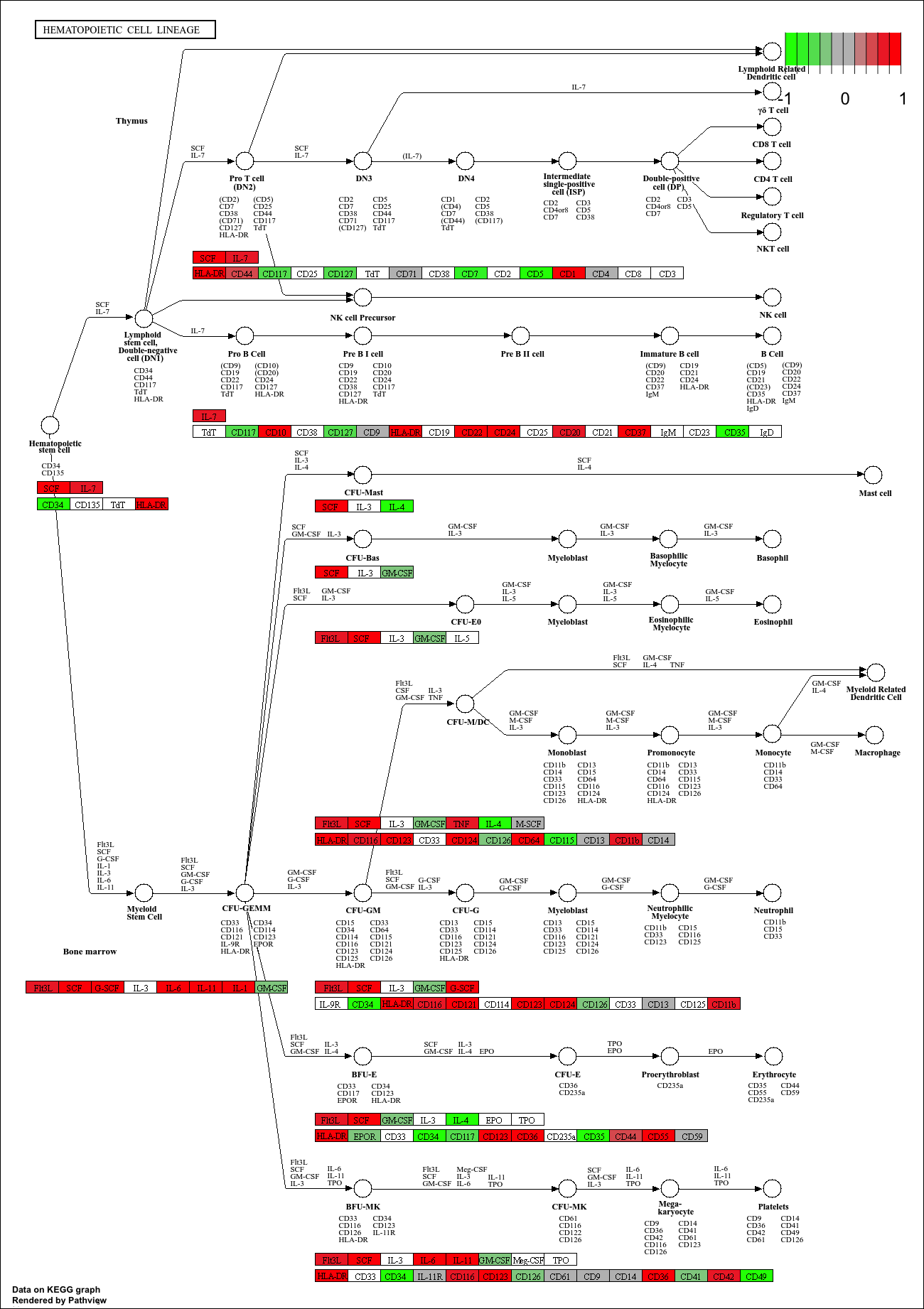
library(gage)
library(gageData)
data(kegg.sets.hs)
data(sigmet.idx.hs)
# Focus on signaling and metabolic pathways only
kegg.sets.hs = kegg.sets.hs[sigmet.idx.hs]
# Examine the first 3 pathways
head(kegg.sets.hs, 3)
$`hsa00232 Caffeine metabolism`
[1] "10" "1544" "1548" "1549" "1553" "7498" "9"
$`hsa00983 Drug metabolism - other enzymes`
[1] "10" "1066" "10720" "10941" "151531" "1548" "1549" "1551"
[9] "1553" "1576" "1577" "1806" "1807" "1890" "221223" "2990"
[17] "3251" "3614" "3615" "3704" "51733" "54490" "54575" "54576"
[25] "54577" "54578" "54579" "54600" "54657" "54658" "54659" "54963"
[33] "574537" "64816" "7083" "7084" "7172" "7363" "7364" "7365"
[41] "7366" "7367" "7371" "7372" "7378" "7498" "79799" "83549"
[49] "8824" "8833" "9" "978"
$`hsa00230 Purine metabolism`
[1] "100" "10201" "10606" "10621" "10622" "10623" "107" "10714"
[9] "108" "10846" "109" "111" "11128" "11164" "112" "113"
[17] "114" "115" "122481" "122622" "124583" "132" "158" "159"
[25] "1633" "171568" "1716" "196883" "203" "204" "205" "221823"
[33] "2272" "22978" "23649" "246721" "25885" "2618" "26289" "270"
[41] "271" "27115" "272" "2766" "2977" "2982" "2983" "2984"
[49] "2986" "2987" "29922" "3000" "30833" "30834" "318" "3251"
[57] "353" "3614" "3615" "3704" "377841" "471" "4830" "4831"
[65] "4832" "4833" "4860" "4881" "4882" "4907" "50484" "50940"
[73] "51082" "51251" "51292" "5136" "5137" "5138" "5139" "5140"
[81] "5141" "5142" "5143" "5144" "5145" "5146" "5147" "5148"
[89] "5149" "5150" "5151" "5152" "5153" "5158" "5167" "5169"
[97] "51728" "5198" "5236" "5313" "5315" "53343" "54107" "5422"
[105] "5424" "5425" "5426" "5427" "5430" "5431" "5432" "5433"
[113] "5434" "5435" "5436" "5437" "5438" "5439" "5440" "5441"
[121] "5471" "548644" "55276" "5557" "5558" "55703" "55811" "55821"
[129] "5631" "5634" "56655" "56953" "56985" "57804" "58497" "6240"
[137] "6241" "64425" "646625" "654364" "661" "7498" "8382" "84172"
[145] "84265" "84284" "84618" "8622" "8654" "87178" "8833" "9060"
[153] "9061" "93034" "953" "9533" "954" "955" "956" "957"
[161] "9583" "9615"
foldchanges = res$log2FoldChange
names(foldchanges) = res$entrez
head(foldchanges)
1266 54855 1465 2034 2150 6659
-2.422719 3.201955 -2.313738 -1.888019 3.344508 2.392288
keggres = gage(foldchanges, gsets=kegg.sets.hs)
attributes(keggres)
$names
[1] "greater" "less" "stats"
head(keggres$less)
p.geomean stat.mean p.val
hsa04110 Cell cycle 8.995727e-06 -4.378644 8.995727e-06
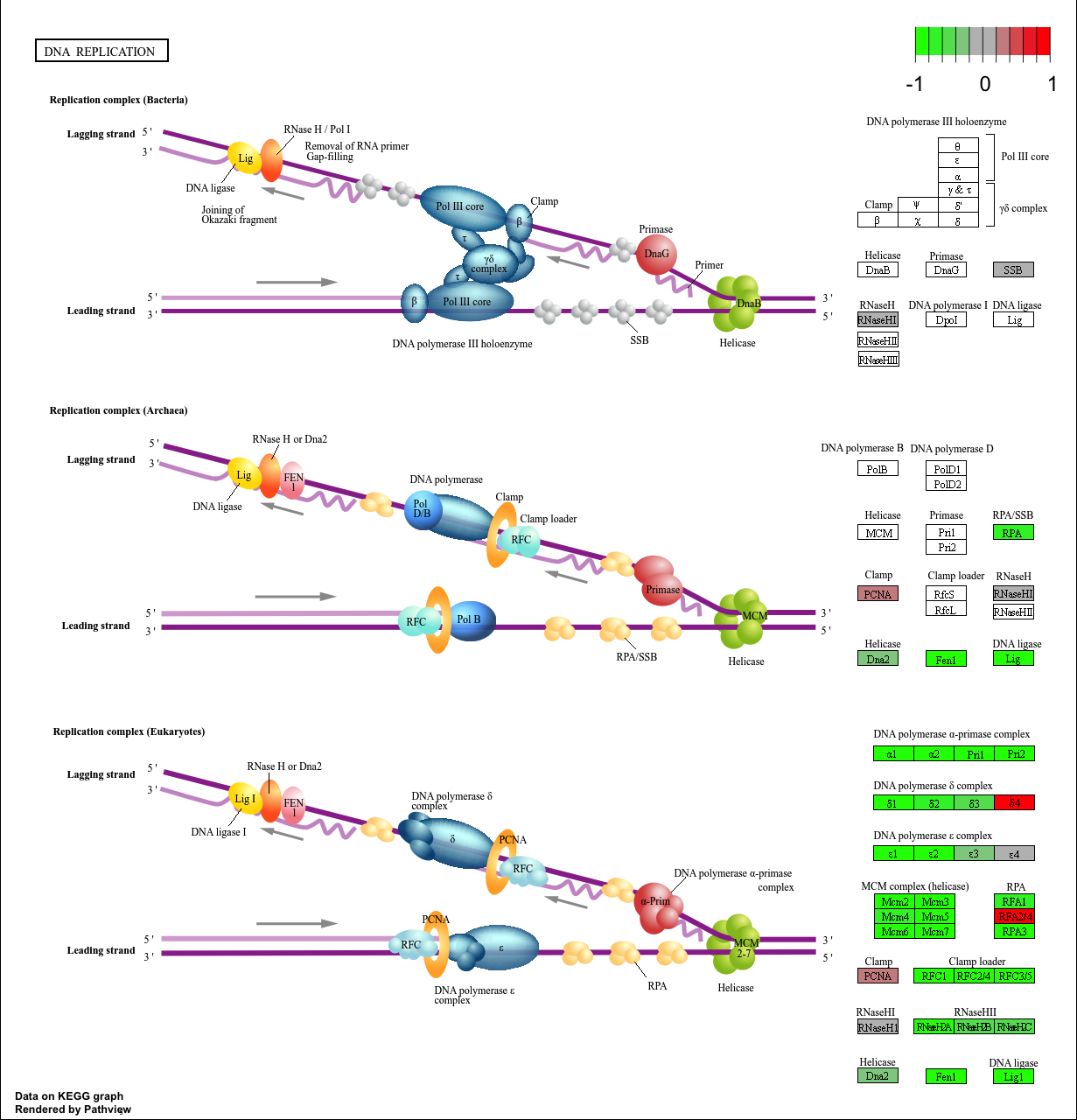
hsa03030 DNA replication 9.424076e-05 -3.951803 9.424076e-05
hsa03013 RNA transport 1.375901e-03 -3.028500 1.375901e-03
hsa03440 Homologous recombination 3.066756e-03 -2.852899 3.066756e-03
hsa04114 Oocyte meiosis 3.784520e-03 -2.698128 3.784520e-03
hsa00010 Glycolysis / Gluconeogenesis 8.961413e-03 -2.405398 8.961413e-03
q.val set.size exp1
hsa04110 Cell cycle 0.001448312 121 8.995727e-06
hsa03030 DNA replication 0.007586381 36 9.424076e-05
hsa03013 RNA transport 0.073840037 144 1.375901e-03
hsa03440 Homologous recombination 0.121861535 28 3.066756e-03
hsa04114 Oocyte meiosis 0.121861535 102 3.784520e-03
hsa00010 Glycolysis / Gluconeogenesis 0.212222694 53 8.961413e-03
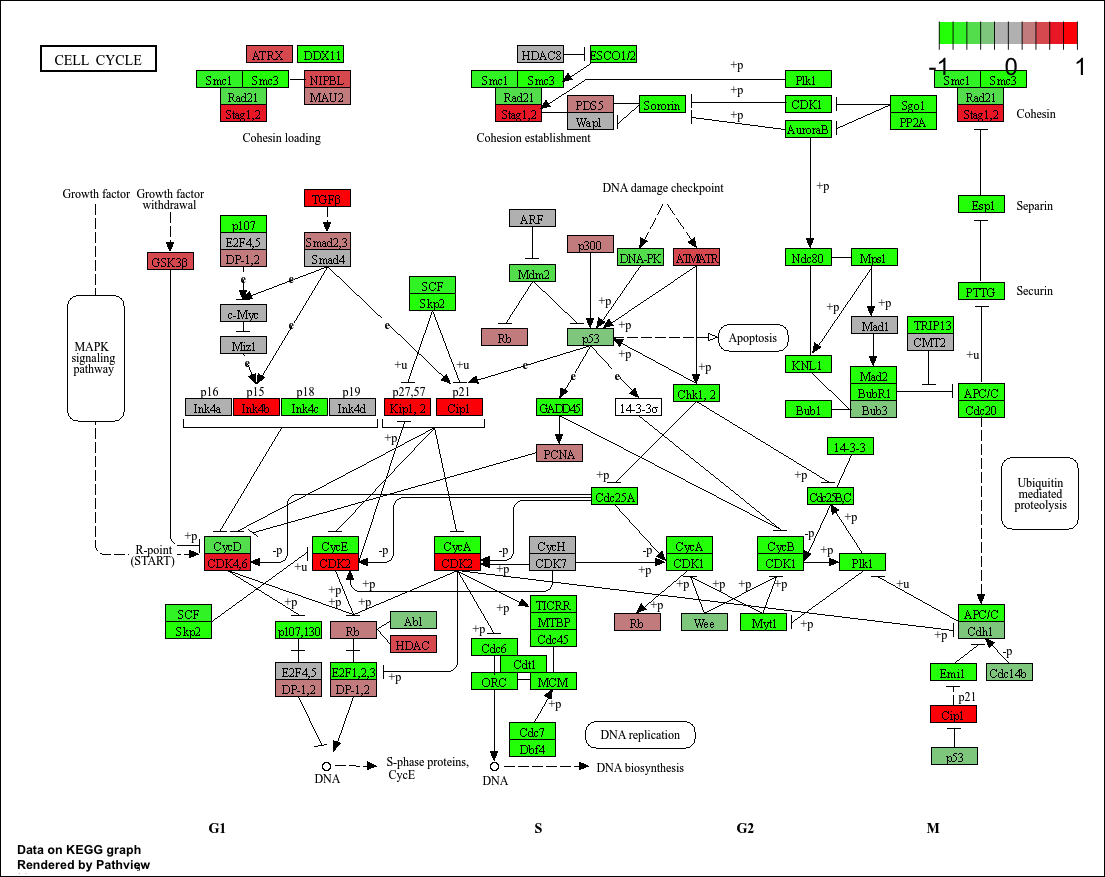
pathview(gene.data=foldchanges, pathway.id="hsa04110")
'select()' returned 1:1 mapping between keys and columns
Info: Working in directory /Users/poojaparthasarathy/Documents/WINTER 2026 UCSD/BIMM143/r wrok/class15/bimm143/class14
Info: Writing image file hsa04110.pathview.png
## Focus on top 5 upregulated pathways here for demo purposes only
keggrespathways <- rownames(keggres$greater)[1:5]
# Extract the 8 character long IDs part of each string
keggresids = substr(keggrespathways, start=1, stop=8)
keggresids
[1] "hsa04640" "hsa04630" "hsa00140" "hsa04142" "hsa04330"
pathview(gene.data=foldchanges, pathway.id=keggresids, species="hsa")
'select()' returned 1:1 mapping between keys and columns
Info: Working in directory /Users/poojaparthasarathy/Documents/WINTER 2026 UCSD/BIMM143/r wrok/class15/bimm143/class14
Info: Writing image file hsa04640.pathview.png
'select()' returned 1:1 mapping between keys and columns
Info: Working in directory /Users/poojaparthasarathy/Documents/WINTER 2026 UCSD/BIMM143/r wrok/class15/bimm143/class14
Info: Writing image file hsa04630.pathview.png
'select()' returned 1:1 mapping between keys and columns
Info: Working in directory /Users/poojaparthasarathy/Documents/WINTER 2026 UCSD/BIMM143/r wrok/class15/bimm143/class14
Info: Writing image file hsa00140.pathview.png
'select()' returned 1:1 mapping between keys and columns
Info: Working in directory /Users/poojaparthasarathy/Documents/WINTER 2026 UCSD/BIMM143/r wrok/class15/bimm143/class14
Info: Writing image file hsa04142.pathview.png
'select()' returned 1:1 mapping between keys and columns
Info: Working in directory /Users/poojaparthasarathy/Documents/WINTER 2026 UCSD/BIMM143/r wrok/class15/bimm143/class14
Info: Writing image file hsa04330.pathview.png
## Focus on lowest 5 upregulated pathwas here for demo purposes only
keggrespathways <- rownames(keggres$less)[1:5]
# Extract the 8 character long IDs part of each string
keggresidsless = substr(keggrespathways, start=1, stop=8)
keggresidsless
[1] "hsa04110" "hsa03030" "hsa03013" "hsa03440" "hsa04114"
pathview(gene.data=foldchanges, pathway.id=keggresidsless, species="hsa")
'select()' returned 1:1 mapping between keys and columns
Info: Working in directory /Users/poojaparthasarathy/Documents/WINTER 2026 UCSD/BIMM143/r wrok/class15/bimm143/class14
Info: Writing image file hsa04110.pathview.png
'select()' returned 1:1 mapping between keys and columns
Info: Working in directory /Users/poojaparthasarathy/Documents/WINTER 2026 UCSD/BIMM143/r wrok/class15/bimm143/class14
Info: Writing image file hsa03030.pathview.png
'select()' returned 1:1 mapping between keys and columns
Info: Working in directory /Users/poojaparthasarathy/Documents/WINTER 2026 UCSD/BIMM143/r wrok/class15/bimm143/class14
Info: Writing image file hsa03013.pathview.png
'select()' returned 1:1 mapping between keys and columns
Info: Working in directory /Users/poojaparthasarathy/Documents/WINTER 2026 UCSD/BIMM143/r wrok/class15/bimm143/class14
Info: Writing image file hsa03440.pathview.png
'select()' returned 1:1 mapping between keys and columns
Info: Working in directory /Users/poojaparthasarathy/Documents/WINTER 2026 UCSD/BIMM143/r wrok/class15/bimm143/class14
Info: Writing image file hsa04114.pathview.png
Gene Ontology
data(go.sets.hs)
data(go.subs.hs)
# Focus on Biological Process subset of GO
gobpsets = go.sets.hs[go.subs.hs$BP]
gobpres = gage(foldchanges, gsets=gobpsets)
lapply(gobpres, head)
$greater
p.geomean stat.mean p.val
GO:0007156 homophilic cell adhesion 8.519724e-05 3.824205 8.519724e-05
GO:0002009 morphogenesis of an epithelium 1.396681e-04 3.653886 1.396681e-04
GO:0048729 tissue morphogenesis 1.432451e-04 3.643242 1.432451e-04
GO:0007610 behavior 1.925222e-04 3.565432 1.925222e-04
GO:0060562 epithelial tube morphogenesis 5.932837e-04 3.261376 5.932837e-04
GO:0035295 tube development 5.953254e-04 3.253665 5.953254e-04
q.val set.size exp1
GO:0007156 homophilic cell adhesion 0.1951953 113 8.519724e-05
GO:0002009 morphogenesis of an epithelium 0.1951953 339 1.396681e-04
GO:0048729 tissue morphogenesis 0.1951953 424 1.432451e-04
GO:0007610 behavior 0.1967577 426 1.925222e-04
GO:0060562 epithelial tube morphogenesis 0.3565320 257 5.932837e-04
GO:0035295 tube development 0.3565320 391 5.953254e-04
$less
p.geomean stat.mean p.val
GO:0048285 organelle fission 1.536227e-15 -8.063910 1.536227e-15
GO:0000280 nuclear division 4.286961e-15 -7.939217 4.286961e-15
GO:0007067 mitosis 4.286961e-15 -7.939217 4.286961e-15
GO:0000087 M phase of mitotic cell cycle 1.169934e-14 -7.797496 1.169934e-14
GO:0007059 chromosome segregation 2.028624e-11 -6.878340 2.028624e-11
GO:0000236 mitotic prometaphase 1.729553e-10 -6.695966 1.729553e-10
q.val set.size exp1
GO:0048285 organelle fission 5.841698e-12 376 1.536227e-15
GO:0000280 nuclear division 5.841698e-12 352 4.286961e-15
GO:0007067 mitosis 5.841698e-12 352 4.286961e-15
GO:0000087 M phase of mitotic cell cycle 1.195672e-11 362 1.169934e-14
GO:0007059 chromosome segregation 1.658603e-08 142 2.028624e-11
GO:0000236 mitotic prometaphase 1.178402e-07 84 1.729553e-10
$stats
stat.mean exp1
GO:0007156 homophilic cell adhesion 3.824205 3.824205
GO:0002009 morphogenesis of an epithelium 3.653886 3.653886
GO:0048729 tissue morphogenesis 3.643242 3.643242
GO:0007610 behavior 3.565432 3.565432
GO:0060562 epithelial tube morphogenesis 3.261376 3.261376
GO:0035295 tube development 3.253665 3.253665
sig_genes <- res[res$padj <= 0.05 & !is.na(res$padj), "symbol"]
print(paste("Total number of significant genes:", length(sig_genes)))
[1] "Total number of significant genes: 8147"
write.table(sig_genes, file="significant_genes.txt", row.names=FALSE, col.names=FALSE, quote=FALSE)
Reactome Result analysis
When comparing these results with KEGG, it is evident that KEGG’s top hit was also Cell Cycle (hsa04110), and Reactome is showing Cell Cycle, Cell Cycle Checkpoints, Mitotic Prometaphase pathways. Both methods are point to the same biology — loss of HOXA1 disrupts cell cycle progression. Differences may come up from the way pathways and gene names seem to be flagged. For instance in this scenario multiple cell cycling related pathways are mapped individually whilst KEGG aggregates them under a more seemingly brad umbrella. Furthermore how gene names may be curated anf flagged could cause differences between how these pathways may be annotated.